Zwei neue Impfreaktionen wurden nach der Corona-Impfung beobachtet: die Immunthrombozytopenie und das Kapillarlecksyndrom. Neue Daten und drei Fallbeispiele.
[wpedon id=3215] Wenn ihr unsere Arbeit unterstützen wollt freuen wir uns
M.Sc. Sharin Santhiraraja-AbreschDocCheck Team
Das Pharmacovigilance Risk Assessment Comittee (PRAC) der EMA ist kürzlich zu dem Schluss gekommen, dass Personen, die vom Kapillarlecksyndrom (CLS) betroffen waren, nicht mit dem Impfstoff Vaxzevria (ursprünglich Astrazeneca) geimpft werden sollten. Ferner soll auch CLS zu den Produktinformationen als neue Nebenwirkung der Vakzine hinzugefügt werden. Bisher wurde jedoch lediglich von 14 Fällen berichtet.
Bei CLS handelt es sich um ein recht seltenes, aber potentiell lebensbedrohliches Krankheitsbild. Es zeichnet sich durch Blutdruckabfall, Hypovolämie und ausgedehnte Ödeme aus. Medizinisches Personal solle demnach auf die Symptome von CLS und dessen Wiederauftreten in Patienten achten, heißt es in der Mitteilung. Personen, die mit Vaxzevria geimpft wurden, sollten sofort einen Arzt aufsuchen, wenn sie in den Tagen nach der Impfung ein schnelles Anschwellen der Arme und Beine oder eine plötzliche Gewichtszunahme bemerken. Diese Symptome sind aufgrund des niedrigen Blutdrucks häufig mit einem Ohnmachtsgefühl verbunden.
ITP-Risiko: Das auch noch
Offenbar ist CLS nicht als einziges Risiko mit einer Vaxzevria-Impfung von Astrazeneca verbunden. Auch das idiopathische thrombozytopenische Purpura (ITP) wird nicht nur mit dem Vektor-Impfstoff assoziiert, sondern auch mit dem mRNA-Impfstoff von Biontech/Pfizer, so eine aktuelle Studie, die im Nature Medicine veröffentlicht wurde.
Bei ITP handelt es sich um eine Thrombozytopenie, die aufgrund einer Autoimmunreaktion gegen Thrombozyten und Megakaryozyten entstanden ist. Die Forschungsergebnisse weisen ein relatives Risiko von 5,77 auf mit einer geschätzten Inzidenz von 1,13 Fällen pro 100.000 Impfdosen. Zwar ist das Risiko solcher Nebenwirkungen recht selten aber vergleichbar mit dem anderer Impfungen wie etwa gegen Hepatitis B, Mumps-Masern-Röteln und Influenza, so eine Mitteilung der University of Edingburgh. Auch über venöse und aterielle Thromboembolien und hämorrhagische Ereignisse wurde berichtet, wobei die Inzidienz bei den Probanden ähnlich ausfiel.
CLS nicht nur bei Astra
Im Annals of Internal Medicine wurde kürzlich von 3 Fallbeispielen berichtet, bei denen die Personen nach einer kürzlich erhaltenen Impfung gegen COVID-19 von CLS betroffen waren. Jedoch wurden diese Ereignisse von der WHO als nicht dosisabhängige und unerwartete Ereignisse klassifiziert. Die 3 Patienten wiesen in ihrer Anamnese bereits auf CLS hin, sodass 1 bis 2 Tage nach Erhalt der COVID-19-Impfung lebensbedrohliche Schübe entwickelt wurden. Die Patienten erhielten entweder Impfstoffe von Johnson & Johnson, Moderna oder Biontec/Pfizer.
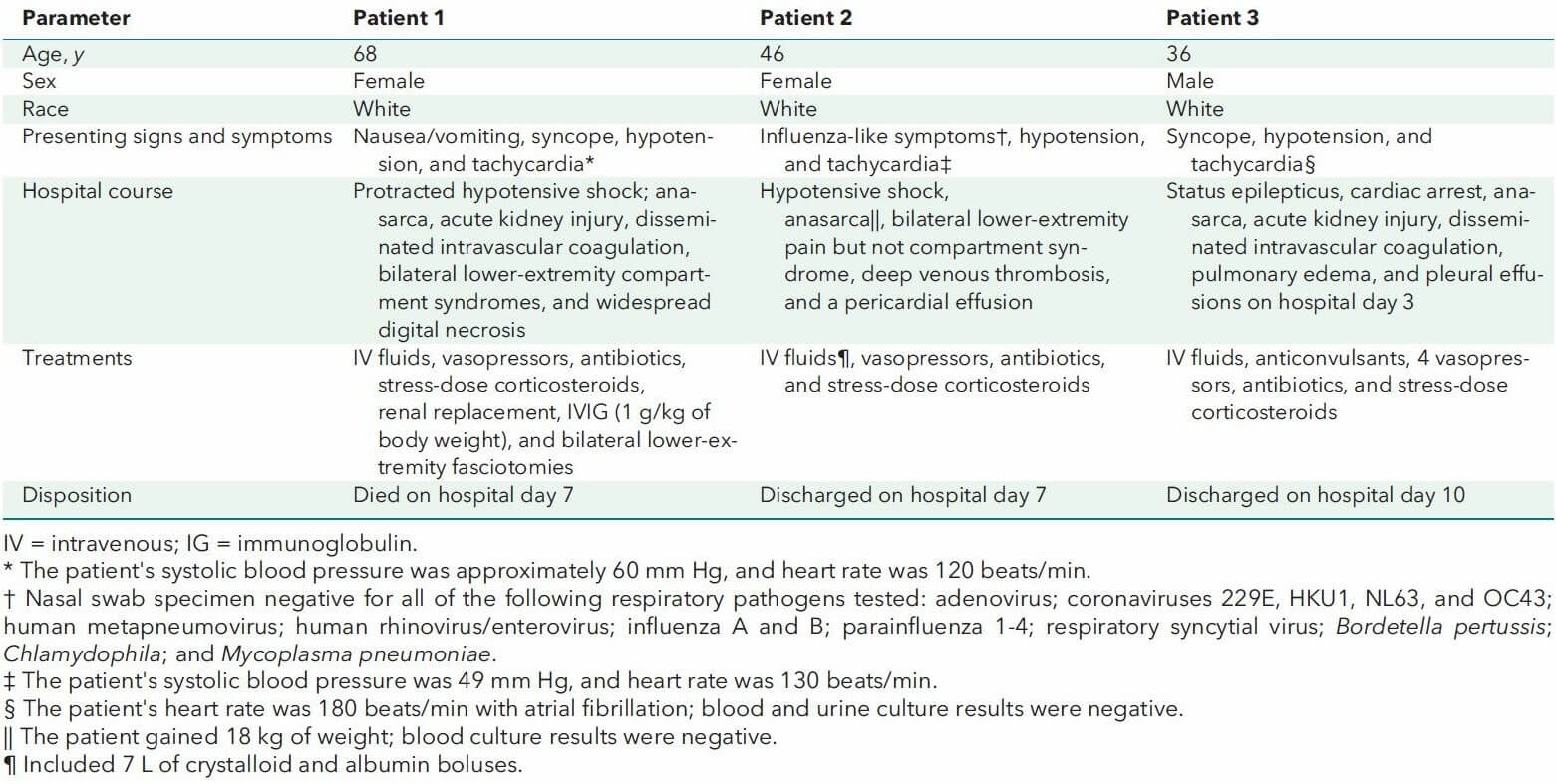 Patienteninformation und hospitalisierte Behandlung der 3 Fallbeispiele. Credit: Matheny et al. (2021)
Patienteninformation und hospitalisierte Behandlung der 3 Fallbeispiele. Credit: Matheny et al. (2021)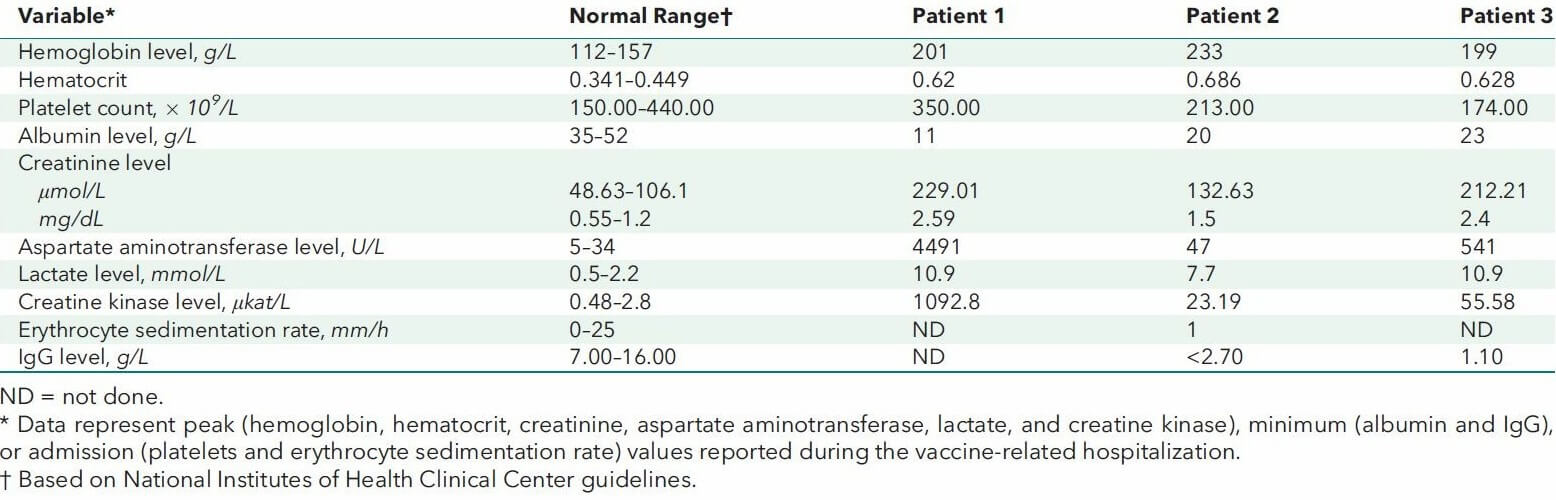 Laborgeprüfte Abnormalitäten in den 3 Fallbeispielen im Vergleich zu den Richtwerten. Credit: Matheny et al. (2021).
Laborgeprüfte Abnormalitäten in den 3 Fallbeispielen im Vergleich zu den Richtwerten. Credit: Matheny et al. (2021).
Die Autoren gehen davon aus, dass CLS als Risikofaktor für die Entwicklung schwerwiegender Nebenwirkungen nach einer COVID-19-Impfung identifiziert werden kann. Jedoch schließen sie andere mögliche Ursachen für das Aufflackern dieser Symptomatik nicht aus, auch wenn kein anderer Auslöser bestimmt werden konnte.
Vergesst nicht die IVIG-Prophylaxe
Laut den Autoren sei eine Entwicklung dieses schubartigen Krankheitsbildes nach einer COVID-19-Impfung ohne CLS-Vorgeschichte unwahrscheinlich. Einige Patienten mit unerklärlichen Episoden von Hypotonie und Ödemen können jedoch nicht diagnostiziertes CLS haben. Zudem erhielten keine der drei vorgestellten Fallbeispiele eine IV-Immunglobulin-Prophylaxe (IVIG-Prophylaxe), als sie geimpft wurden. Im Kontrast dazu wurden in den anderen 78 geimpften CLS-Patienten, die zuvor eine IVIG-Prophylaxe erhielten, keine CLS-Schübe diagnostiziert. Daher wird bei einer CLS-Diagnose oder Verdachtsdiagnose eine IVIG-Prophylaxe vor Erhalt der Impfung empfohlen.
Diese Fallbeispiele sind in der Praxis wichtig, insbesondere bei neuartigen Krankheitsbildern die mit einer COVID-19-Infektion oder einer Impfung entstehen können. Dadurch wird schon im Vorfeld auf mögliche Komplikationen aufmerksam gemacht, die von dem behandelnden medizinischen Personal zu beachten sind. Zudem zeigt sich, dass nicht nur bei einer Impfung mit Astrazeneca auf diese spezifische Symptomatik und weitere Nebenwirkungen geachtet werden muss, sondern auch bei den beliebten mRNA-Impfstoffen.
Quelle
Definition Kappilarlecksyndrom
https://flexikon.doccheck.com/de/Clarkson-Syndom
1 Definition
Beim sog. Kapillarlecksyndrom, kurz CLS, handelt es sich um ein seltenes, potentiell lebensbedrohliches Krankheitsbild, bei dem es zu einem Austritt von Blutplasma und Plasmaproteinen aus den Kapillargefäßen in das Interstitium kommt. Es zeichnet sich durch Blutdruckabfall, Hypovolämie und ausgedehnte Ödeme aus.
2 Epidemiologie
Seit der Erstbeschreibung des Krankheitsbildes im Jahre 1960 wurden weltweit nur wenige Dutzend Fälle beschrieben. Das mittlere Erkrankungsalter liegt bei 45 Jahren.
3 Pathophysiologie
Die genauen Pathomechanismen des Kapillarlecksyndroms sind zurzeit (2021) noch unklar. Zytokine wie Interleukin-2 scheinen eine wichtige Rolle zu spielen. Weiterhin ist auffällig, dass ein großer Teil der Patienten (> 80%) an einer monoklonalen Gammopathie unklarer Signifikanz (MGUS) leidet.
Manchmal wird ein Kapillarlecksyndrom durch Arzneistoffe ausgelöst, u.a. durch Gemcitabin, Tagraxofusp, Tisagenlecleucel und Axicabtagen-Ciloleucel sowie Interleukine und monoklonale Antikörper.
4 Klinik
Charakteristischerweise ist die Erkrankung gekennzeichnet durch das Abwechseln von Krisen und Ruhephasen. Dabei kann es im Laufe des Lebens lediglich zu einer einzelnen Krise kommen, die Episoden können aber auch mehrmals pro Jahr auftreten. Trigger solcher Krisen können Atemwegsinfekte sein. Die Erkrankung kann aber auch perimenstruell oder postpartal auftreten. Selten ist intensiver Sport zeitlich korreliert.
Klinisch lassen sich die Krisen in vier Schweregrade einteilen, die von einer Hypotension, die auf orale Flüssigkeitszufuhr anspricht, bis zum tödlichen hypovolämischen Schock reichen. Typisch ist ein dreiphasiger Verlauf aus Prodromalphase, Leckphase und Post-Leckphase.
Definition Idiopathische thrombozytopenische Purpura
https://flexikon.doccheck.com/de/ITP
1 Definition
Unter der Immunthrombozytopenie, kurz ITP, versteht man eine erworbene Thrombozytopenie aufgrund einer Autoimmunreaktion gegen Thrombozyten und Megakaryozyten. Dabei liegt die Thrombozytenzahl per definitionem wiederholt unter 100.000/µl.
- ICD10-Code: D69.3
2 Nomenklatur
Der frühere Begriff idiopathische thrombozytopenische Purpura sollte nicht mehr verwendet werden, da die ITP aufgrund der Erkenntnisse zur ursächlichen Immundysregulation nicht als idiopathisch bezeichnet werden kann. Außerdem tritt eine Purpura nicht bei allen ITP-Patienten auf.
3 Epidemiologie
Die Inzidenz der Immunthrombozytopenie liegt bei Erwachsenen bei etwa 0,2-0,4 Neuerkrankungen pro 10.000 Einwohner pro Jahr. Die Prävalenz beträgt 0,9-2,6 pro 10.000.
Bei Kindern und Jugendlichen beträgt die Inzidenz 0,2-0,7/10.000/Jahr und die Prävalenz 0,4-0,5/10.000. Die geringere Prävalenz liegt darin begründet, dass die ITP bei Kindern nur in 20-30 % d.F. chronisch verläuft; bei Erwachsenen hingegen in 60 %.
Das mittlere Erkrankungsalter beträgt zwischen 50-55 Jahren, wobei zunehmend ältere Personen an einer ITP erkranken. Mädchen und Frauen sind grundsätzlich häufiger betroffen, ab dem 60. Lebensjahr überwiegt dann jedoch das männliche Geschlecht.
Bei Kindern tritt eine akut verlaufende ITP häufiger im Frühjahr auf, vermutlich da sie oft durch Infekte im Winter getriggert wird.
4 Ätiologie
Bei der ITP unterscheidet man zwischen zwei Formen:
| Form | Ursache |
|---|---|
| Primäre ITP (80%) | keine auslösende Ursache erkennbar |
| Sekundäre ITP (20%) | Autoimmunerkrankungen: Antiphospholipid-Syndrom, Sjögren-Syndrom, systemischer Lupus erythematodes, rheumatoide Arthritis, Autoimmunthyreoiditis Chronisch entzündliche Darmerkrankungen: Colitis ulcerosa, Morbus Crohn Hämatologische Neoplasien: Myelodysplastische Syndrome, Lymphome (v.a. chronische lymphatische Leukämie) Solide Malignome: v.a. Bronchial- und Mammakarzinome Immundefizienz-Syndrome (z.B. CVID, Canale-Smith-Syndrom, Wiskott-Aldrich-Syndrom) Allogene Stammzelltransplantation Virusinfektionen (CMV-, HIV-Infektion, Hepatitis B, Hepatitis C) Medikamente (z.B. NSAR, Ampicillin, Sulfonamide, Carbamazepin) Impfungen |
Das gleichzeitige Vorliegen einer ITP und einer autoimmunhämolytischen Anämie wird als Evans-Syndrom bezeichnet.
Eine ITP ist weiterhin mit einer Helicobacter-pylori-Infektion der Magenschleimhaut assoziiert, insbesondere bei Patienten in Asien. Die pathogenetische Bedeutung ist derzeit (2020) unklar, trotzdem sollten erwachsene ITP-Patienten auf Helicobacter untersucht werden und ggf. eine Eradikationstherapie erhalten.
5 Pathogenese
Die ITP ist eine erworbene Thrombozytopenie, die durch eine Autoimmunreaktion gegen Thrombozyten und Megakaryozyten entsteht. Dabei spielen verschiedene Pathomechanismen eine entscheidende Rolle:
- Autoantikörper gegen Thrombozyten
- Immundysregulation
- Hemmung der Thrombozytopoese
5.1 Thrombozytenantikörper
Patienten mit ITP besitzen Autoantikörper gegen Oberflächenproteine von Thrombozyten (z.B. Glykoprotein Ib/IX oder Glykoprotein IIb/IIIa). Diese Antikörper-markierten Thrombozyten binden an Fc-Rezeptoren von Makrophagen und dendritischen Zellen in Milz und Leber und werden vermehrt abgebaut. Außerdem können die Antikörper Thrombozyten über eine Komplementaktivierung schädigen. Geschädigte Thrombozyten binden vermehrt an Ashwell-Morell-Rezeptoren in der Leber und werden abgebaut.
Des Weiteren können die Autoantikörper auch zur Thrombozytopathie führen, was eine verstärkte Blutungsneigung zur Folge hat.
5.2 Immundysregulation
Bei der ITP findet sich eine verminderte Zahl an regulatorischen T-Lymphozyten (TREGs), sodass eine Immundysregulation die Folge ist. Die Ursache dieses Ungleichgewichts ist derzeit (2020) unklar. Möglicherweise spielen mesenchymale Stammzellen hierbei eine Rolle, die bei ITP-Patienten vermindert und funktionsgestört sind. T-Lymphozyten können weiterhin direkt Thrombozyten und Megakaryozyten schädigen.
5.3 Hemmung der Thrombozytopoese
Autoantikörper schädigen nicht nur Thrombozyten, sondern auch Megakaryozyten. Hierbei binden die Antikörper insbesondere an GP Ib/IX. Auch Antikörper gegen Thrombopoietin können vorkommen. Daneben findet sich häufig ein relativer Thrombopoietinmangel, also ein nicht ausreichend hoher Spiegel, vermutlich weil Thrombopoietin an Thrombozyten bindet und folglich vermehrt abgebaut wird. Auch die reaktiv erhöhte Zahl an Megakaryozyten im Knochenmark führt zur Bindung von Thrombopoietin. Zuletzt führen Antikörper gegen Glykoprotein Ib zur reduzierten Thrombopoietinsynthese in der Leber.
6 Klinik
Eine ITP manifestiert sich primär in Form einer erhöhten Blutungsneigung.
6.1 Blutungen
Typische blutungsbedingte Symptome bei ITP-Patienten sind:
- nicht palpable Petechien an den Beinen, seltener am Rumpf und an den Armen
- Blutungen der Schleimhäute von Mund und Nase
- urogenitale Blutungen, Hypermenorrhoe
- verstärkte Blutungen und Hämatomneigung bei kleinen Traumen
- selten innere Blutungen (z.B. intrazerebrale Blutung)
Eher untypisch sind flächenhafte Hämatome (Ekchymosen bzw. Sugillationen) sowie Gelenkblutungen.
Zum Zeitpunkt der Diagnosestellung haben 10 % der pädiatrischen und 20-30 % der erwachsenen Patienten keine Blutungssymptome. Im chronischen Stadium finden sich Blutungssymptome bei 60-70 % der Patienten. Grundsätzlich ist die Blutungsneigung bei ITP relativ gering, im Vergleich zu einer vergleichbaren Thombozytopenie anderer Ursache.
Als Folge des vermehrten Blutverlustes kann es weiterhin zu Symptomen einer Eisenmangelanämie kommen.
6.2 Weitere Symptome
ITP-Patienten haben ein erhöhtes Infektionsrisiko, da Thrombozyten auch eine Rolle bei der Infektabwehr spielen. Außerdem beklagen viele Betroffene Erschöpfungssymptome, Fatigue, kognitive Funktionseinschränkungen und depressive Störungen. Die Ursache hierfür ist derzeit (2020) unklar und vermutlich mutlifaktoriell bedingt (Einschränkungen der körperlichen Aktivität und der sozialen Teilhabe, Schlafstörungen etc.)
6.3 Thromboembolien
Eine ITP führt zu einem zweifach erhöhten Risiko von venösen und arteriellen Thromboembolien. Ausgenommen sind Patienten mit einer Thrombozytenzahl < 50.000/µl. Der Grund für die Thrombophilie ist derzeit (2020) unklar. Besonders hoch ist das Risiko:
- nach Splenektomie
- nach bereits stattgehabter Thromboembolie
- bei schnellem Thrombozytenanstieg unter Therapie
- bei älteren Patienten, Adipositas und Operationen
- bei Vorliegen von Antiphospholipid-Antikörpern oder Lupus-Antikoagulans
7 Diagnostik
Bei der ITP handelt es sich um eine Ausschlussdiagnose. Sie manifestiert sich labordiagnostisch in Form einer Thrombozytopenie (bei der ITP als < 100.000/µl definiert) bei sonst normalem peripherem Blutbild und Blutausstrich.
7.1 Basisdiagnostik
Zur Basisdiagnostik sollten folgende Aspekte evaluiert werden:
- Anamnese: Blutungen, Infektionen, Medikamente, Alkohol, Schwangerschaft, Thrombosen, Familien- und Berufsanamnese
- Körperliche Untersuchung: Blutungszeichen, Lymphknotenstatus, Leber- und Milzgröße, Exantheme usw.
- Blutbild: Ausschluss einer Pseudothrombozytopenie
- Blutausstrich
- Gerinnungsparameter: Quick- bzw. INR-Wert, aPTT, Fibrinogen
- Blutzucker
- Stuhluntersuchung auf Blut (z.B. iFOBT), ggf. Gastroduodenoskopie zum Ausschluss einer oberen GI-Blutung
- Knochenmarkdiagnostik: bei atypischen Befunden sowie bei Patienten über 60 Jahre zum Ausschluss von Differenzialdiagnosen. Bei der ITP zeigen sich vermehrt junge Megakaryozyten bei auch insgesamt vermehrten Megakaryozyten.
- Untersuchung auf irreguläre erythrozytäre Autoantikörper: v.a. bei gleichzeitiger Anämie (Evans-Syndrom)
7.2 Weitergehende Diagnostik
- Serumelektrophorese, Serumimmunglobuline: Ausschluss von Immundefektsyndromen sowie eines multiplen Myeloms
- Autoimmundiagnostik: CCP-Antikörper, ANA, ANCA, Anti-dsDNA, Antiphospholipid-Antikörper, Lupus-Antikoagulans
- Thrombozyten-Autoantikörper: Bestimmung nur bei persistierender oder chronischer ITP und atypischem Krankheitsverlauf, wenn Zweifel an der Diagnose einer ITP bestehen. Relevant ist nur der Nachweis von an Thrombozyten gebundenen Autoantikörpern gegen GP Ib/IX und IIb/IIIa. Freie Autoantikörper im Serum haben keine diagnostische Bedeutung. Bei negativem Befund ist eine ITP nicht ausgeschlossen.
- von-Willebrand-Faktor-Multimeranalyse
- Schilddrüsendiagnostik
- Diagnostik auf Helicobacter pylori
- Hepatitis B-, C- und HIV-Serologie
- Bildgebende Verfahren bei Verdacht auf hämatologische oder solide Tumoren. Eine Splenomegalie kann bei ITP vorkommen, dabei sollten jedoch z.B. Lymphome oder ein Morbus Gaucher ausgeschlossen werden.
7.3 Stadien
Die Einteilung in Stadien ist von therapeutischer Relevanz. Man unterscheidet in Abhängigkeit von der Diagnosestellung (und unabhängig vom Symptombeginn):
- neu diagnostizierte ITP: bis zu 3 Monate nach Diagnosestellung, noch häufige Spontanremission
- persistierende ITP: zwischen 3-12 Monate nach Diagnosestellung, Spontanremissionen sind weniger häufig
- chronische ITP: > 12 Monate nach Diagnosestellung, Spotanremission eher unwahrscheinlich
7.4 Blutungsscores
Zur Einschätzung der klinischen Blutungsneigung wird bei ITP-Patienten eine Stratifizierung entsprechend der Blutungsgrade nach WHO vorgenommen:
| WHO-Blutungsgrad | Beschreibung |
|---|---|
| 0 | keine Blutungszeichen |
| I | Petechien, kleine Hämatome, Ekchymosen (< 10 cm) Schleimhautblutungen (Mund, Nase) Epistaxis (< 1 Stunde Dauer, keine Notwendigkeit einer ärztlichen Intervention) subkonjunktivale Blutungen vaginale Blutungen (unabhängig von Menstruation, ≤ 2 Binden/Tag notwendig) |
| II | Hämatome, Ekchymosen (> 10 cm) Epistaxis (> 1 Stunde Dauer oder Tamponade notwendig) retinale Blutungen ohne Visusminderung vaginale Blutungen (unabhängig von Menstruation, > 2 Binden/Tag notwendig) Melaena, Hämatemesis, Hämoptysen, Hämaturie, Hämatochezie Blutungen aus Punktionsstellen Blutungen in Muskeln und Gelenke |
| III | wie II nur transfusionspflichtig |
| IV | retinale Blutungen mit Visusminderung ZNS-Blutungen andere Organblutungen, welche die Organfunktion gefährden letale Blutungen |
Für pädiatrische Patienten eignet sich der modifizierte Buchanan-Score:[1]
| Grad | Beschreibung |
|---|---|
| 0 | keine frischen Blutungszeichen |
| 1 | wenig Petechien (< 100) und/oder < 5 kleine Hämatome (< 3 cm) keine Schleimhautblutungen |
| 2 | viele Petechien und > 5 große Hämatome (> 3 cm) |
| 3a | Mundschleimhautblutungen Blutkrusten in Nasenlöchern milde Epistaxis (< 5 Minuten) |
| 3b | Epistaxis > 5 Minuten Makrohämaturie rektale Blutungen schmerzhafte Mundschleimhautblutungen signifikante Menorrhagie |
| 4 | Schleimhautblutungen oder Blutungen innerer Organe (Gehirn, Lunge, Muskulatur, Gelenke) mit Notwendigkeit zur umgehenden medizinischen Versorgung oder Intervention |
| 5 | nachgewiesene intrakranielle Blutung lebensbedrohliche bzw. tödliche Blutung jeder Lokalisation |
7.5 TH2-Score
Der TH2-Score dient der Beurteilung des Thrombose- und Blutungsrisikos, um festzustellen, ob der ITP-Patient ggf. eine Antikoagulation benötigt:
| CHA2DS2-VASc-Score > 5 oder spontane oder rezidivierte oder tumorassoziierte venöse Thromboembolie oder Antiphospholipid-Syndrom | + 1 |
| Therapie mit IVIG in letzten 2 Wochen oder Therapie mit Thrombopoietin-Rezeptor-Agonisten (TRA) in letzten 4 Wochen oder Splenektomie in letzten 4 Wochen | + 1 |
| Thrombozyten < 20.000/µl | – 1 |
| Blutung WHO ≥ 2° vor Therapiebeginn | – 1 |
Bei einem TH2-Score von 0-2 ist das Thromboserisiko höher, bei Werten von -2 bis -1 überwiegt das Blutungsrisiko.
8 Differenzialdiagnosen
Die ITP muss von anderen Ursachen einer Thrombozytopenie abgegrenzt werden. Beispielsweise kann pathogenetisch eine verminderte Thrombozytenbildung zu Grunde liegen. Mögliche Ursachen dafür sind:
- Schädigung des Knochenmarks durch Medikamente, Alkohol oder Zytostatika
- Infiltration und Verdrängung des Knochenmarks durch hämatologische oder solide Neoplasien
- Myelofibrose
- Myelodysplastische Syndrome
- Paroxysmale nächtliche Hämoglobinurie (PNH)
- schwerer Vitaminmangel
- genetische Defekte (z.B. Bernard-Soulier-Syndrom, MYH9-assoziierte Syndrome, Wiskott-Aldrich-Syndrom)
Immunologisch bedingte Thrombozytopenien finden sich auch nach Einnahme von Heparin (HIT) oder Glykoprotein-IIb/IIIa-Rezeptorantagonisten sowie bei posttransfusioneller Purpura, Schwangerschafts-assoziierter Thrombozytopenie sowie der neonatalen Alloimmunthrombozytopenie (NAIT).
Nicht immunologisch bedingte Ursachen eines erhöhten Thrombozytenverbrauchs finden sich bei mikroangiopathischen hämolytischen Anämien (z.B. TTP, HUS), Verbrauchskoagulopathie, Willebrand-Jürgens-Syndrom Typ IIb, massiver Lungenembolie, großen Hämangiomen (z.B. Kasabach-Merritt-Syndrom) und Aneurysmen.
Weitere Formen der Thrombozytopenien treten im Rahmen einer Splenomegalie (Hypersplenismus), bei massiver Blutung und bei schweren Infektionen auf.
Zuletzt muss auch an eine Pseudothrombozytopenie gedacht werden.
Bei Vorliegen von palpablen Petechien muss eine Vaskulitis erwogen werden. Flächenhafte Hämatome oder Gelenkblutungen lassen an eine plasmatische Gerinnungsstörung (z.B. Hämophilie A oder B) denken. Die wichtigste Differenzialdiagnose einer neu-diagnostizierten ITP im Kindesalter ist eine akute lymphatische Leukämie (ALL).
| ITP | TTP | DIC |
|---|---|---|
| IgG-Antikörper gegen Thrombozyten | Endotheldefekt | Überschuss/Aktivierung von Thrombin |
| INR und PTT normal | INR und PTT normal | INR und PTT erhöht |
| Fibrinogen und D-Dimer normal | Fibrinogen und D-Dimer normal | Fibrinogen erniedrigt, D-Dimer erhöht |
| weniger symptomatisch | stärker symptomatisch | stärker symptomatisch |
9 Therapie
Die Therapie der ITP ist abhängig von der Blutungsneigung, der Thrombozytenzahl, dem Krankheitsstadium und -verlauf sowie von weiteren individuellen Faktoren. Es existiert kein Schwellenwert der Thrombozytenzahl, bei dessen Unterschreiten eine Behandlung grundsätzlich indiziert ist.
Außerdem sollten Einschränkungen der Lebensqualität erfasst und auf Fatigue-Symptome geachtet werden. Patienten mit ITP können Sport machen; bei Thrombozytenzahlen < 50.000/µl sollten Kampf- und Kontaktsportarten vermieden werden.
9.1 Erstlinientherapie
Eine Therapie ist bei folgenden Patienten mit neu-diagnostizierter ITP zu erwägen:
- WHO-Blutungsneigung 0° bis II° und Thrombozytenwerten < 20.000-30.000/µl
- WHO-Blutungsneigung III° bis IV°
In anderen Fällen kann auch abgewartet werden (Watchful Waiting).
9.1.1 Glukokortikoide
Zur Erstlinienthereapie eignen sich Glukokortikoide. Sie wirken immunsuppressiv und sollen die Bildung der Autoantikörper hemmen. Nach Absetzen der Steroide fallen die Thrombozytenwerte jedoch häufig wieder ab, sodass dauerhafte Remissionen eher selten sind.
Die Therapiedauer sollte mindestens drei Monate betragen, wobei deutlich längere Therapien keine Verbesserung der Ansprechrate erbringen. Außerdem müssen vorbeugende Maßnahmen zur Reduktion von Nebenwirkungen beachtet werden (z.B. Osteoporoseprophylaxe).
Bevorzugt wird Dexamethason eingesetzt, jedoch ist auch die Gabe von Prednisolon oder Methylprednisolon möglich.
Pädiatrischen Patienten mit chronischer ITP sollten in speziellen Zentren betreut werden, da bisher kein Therapiestandard definiert ist. Zur Erstlinientherapie werden ebenfalls häufig Glukokortikoide eingesetzt, jedoch kann in vielen Fällen auf eine Therapie verzichtet werden.
9.1.2 Weitere Therapieoptionen
Bei schweren oder lebensbedrohlichen Blutungen werden zusätzlich intravenöse Immunglobuline (IVIG) verabreicht. Sie blockieren die Phagozytose von antikörperbeladenen Thrombozyten und führen zu einem schnellen Anstieg der Thrombozytenzahl, jedoch ohne eine anhaltende Remission zu erzielen. Anti-D-Immunglobulin führt zur limitierten Hämolyse mit Absättigung der Fc-Rezeptoren durch antikörperbeladene Zellen und bewirkt damit die Hemmung der Fc-Rezeptorfunktion. Es ist jedoch nur bei Rhesus-positiven Patienten mit erhaltener Milzfunktion wirksam. Die in Deutschland erhältlichen Präparate (z.B. Rhophylax®, Rhesonativ®) sind nur für die Prophylaxe des Morbus haemolyticus neonatorum zugelassen. Rozrolimupab, eine Mischung aus 25 rekombinanten monoklonalen Anti-D-Immunglobulinen, ist derzeit (2020) noch nicht zugelassen.[2]
Bei schweren Blutungen können weiterhin Thrombozytenkonzentrate zum kurzfristigen Anstieg der Thrombozytenzahl und Sistieren der Blutung eingesetzt werden. Sie bergen jedoch das Risiko einer Induktion von Auto- oder Alloantikörpern.
Auch der zusätzliche Einsatz von Rituximab oder Thrombopoietin-Rezeptor-Agonisten (TRA) kann notwendig sein. Rituximab induziert eine Depletion von B-Zellen, sodass weniger Autoantikörper gebildet werden. TRA sind nur für die chronische ITP zugelassen, jedoch können auch Patienten mit neu-diagnostizierter oder persistierender ITP im Notfall von der Gabe profitieren. Meist werden die TRA dann mit Glukokortikoiden kombiniert. Romiplostim (Nplate®) ist seit 2009, Eltrombopag (Revolade®) seit 2010 in der EU zugelassen.
In seltenen Fällen ist bei einer schweren Blutung eine Notfallsplenektomie indiziert.
9.2 Zweitlinientherapie
Eine Zweitlinientherapie kann eingeleitet werden, wenn:
- die Erstlinientherapie nach 2-4 Wochen nicht zum Ansprechen führt,
- die Erstlinientherapie schlecht vertragen wird,
- es zu einem Rezidiv innerhalb der ersten 6 Monate kommt.
Dabei ist die Indikation zur Behandlung immer eine individuelle Entscheidung. Kommt es beispielsweise nach 6 Monaten zu einem Rezidiv, kann nochmal eine Erstlinientherapie versucht werden. Eine medikamentöse Zweitlinientherapie kann bei Patienten mit WHO-Blutungsneigung 0° bis II° erwogen werden. Bei schweren Blutungen (III-IV°) besteht immer eine Therapieindikation.
9.2.1 Thrombopoietin-Rezeptor-Agonisten
Thrombopoietin-Rezeptor-Agonisten (TRAs) führen bei über 90 % der Patienten zu einem kurzfristigen, bei 30-90 % der Patienten zu einem langfristigen Ansprechen. Der Zielbereich der Thrombozytenzahl liegt bei 50.000-150.000/µl; eine Normalisierung wird nicht angestrebt. Eltrombopag und Romiplostim sind nicht kreuzresistent, d.h. wenn der eine TRA nicht ausreichend wirksam ist, kann durchaus der andere noch ansprechen. Während Eltrombopag per os appliziert wird, muss Romiplostin subkutan gegeben werden. Die Wirkstoffe besitzen weitgehend ähnliche Nebenwirkungen und unterscheiden sich kaum in der Wirksamkeit.
Nach dem Absetzen von TRAs finden sich anhaltende Remissionen bei 13-30 % der Patienten. Entsprechend kann bei Thrombozytenzahlen, die sich längere Zeit im Zielbereich befinden, ein Absetzversuch unternommen werden, jedoch ist ein langsames Ausschleichen über Monate notwendig.
Weitere bisher (2020) in Europa nicht zugelassene TRAs sind:
- TPIAO (auch rHuTPO genannt): rekombinantes Thrombopoietin-Molekül, das in China seit 2010 zugelassen ist. In Gegensatz zu Romiplostim und Eltrombopag scheint TPIAO bei schwangeren Patientinnen wirksam und sicher zu sein.
- Avatrombopag (Doptelet®): ähnelt Eltrombopag, jedoch verlängerte Halbwertszeit und nahrungsunabhängige Resorption. Seit 2019 zur Therapie der Thrombozytopenie bei chronischer Lebererkrankung vor operativen Eingriffen zugelassen, jedoch nicht für ITP-Patienten.
- Lusutrombopag (Lusutrombopag Shionogi®): ähnelt Avatrombopag, ebenfalls keine Zulassung für die ITP.
9.2.2 Weitere Therapieoptionen
Sprechen Patienten unter TRA-Monotherapie nicht an, kann die Kombination mit einem niedrig dosierten Steroid erwogen werden.
Weitere Optionen der Zweitlinientherapie sind:
- Fostamatinib (Tavalisse®): Seit 2020 ist der SYK-Inhibitor Fostamatinib in Europa zur Zweitlinientherapie für Patienten zugelassen, die eine parenterale Therapie (z.B. Romiplostin) ablehnen und mit den Nahrungsrestriktionen von Eltrombopag nicht zurechtkommen. Fostamatinib reduziert durch Hemmung der Milztyrosinkinase Syk die Antikörper-vermittelte Zerstörung von Thrombozyten.
- Splenektomie: Zwei Drittel der Patienten nach Splenektomie erreichen eine partielle oder komplette Remission. Indikation bei Patienten mit persistierender oder chronischer ITP und schweren Blutungen (III-IV°) ohne ausreichendes Ansprechen auf bisherige Therapien. Aufgrund der verbundenen Risiken nach Splenektomie wird sie eher selten durchgeführt.
- Rituximab: bei Patienten, bei denen eine Splenektomie zwar notwendig, aber nicht möglich oder erwünscht ist.
- Tranexamsäure: bei pädiatrischen Patienten mit Schleimhautblutungen, Menorrhagien oder im Rahmen von Zahneingriffen.
9.3 Drittlinientherapie
Behandlungsmöglichkeiten in der Drittlinientherapie sind:
- Azathioprin
- Ciclosporin
- Cyclophosphamid
- Danazol
- Dapson
- Hydroxychloroquin
- Mycophenolat
- Rituximab: erreicht bei 60 % der Patienten mit chronischer, therapierefraktärer ITP eine kurzfristige Steigerung der Thrombozytenzahl und bei 10-40 % der Patienten eine längerfristige Remission.
- Fostamatinib
- Splenektomie
Die einzelnen Arzneistoffe müssen z.T. off label eingesetzt werden und können auch untereinander kombiniert werden.
10 Prognose
Ungefähr ⅓ bis ⅔ der Patienten mit chronischer ITP erreichen z.T. noch nach mehreren Jahren eine partielle oder komplettte Remission. Dabei gibt es jedoch keinen Marker, der verlässlich einen chronischen Verlauf vorhersagen kann. Für einen selbstlimitierenden Verlauf sprechen:
- Beginn im jungen Alter
- Beginn nach Infekt
- plötzlicher Beginn
- ausgeprägte Blutungsneigung
- gutes Therapieansprechen
Ein chronischer Verlauf findet sich hingegen häufiger bei:
- Erwachsenen (v.a. < 60. Lebensjahr)
- primärer ITP
- schleichendem Beginn
- geringer Blutungsneigung bzw. asymptomatischen Patienten
- fehlender bzw. geringer Ansprache auf Erstlinientherapie
Angaben zum Blutungsrisiko und zur Mortalität basieren meist auf alten Studien und sind aufgrund von veränderten Therapiemaßnahmen wenig aussagekräftig. Die Mortalität liegt in aktuellen pädiatrischen Studien bei 0 %, bei Erwachsenen bei 0-7 %.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hinterlasse einen Kommentar