Bereits 2021 hatten Wissenschaftler die Herausgabe der Zulassungsunterlagen verlangt. In den USA haben Forscher und Wissenschaftler nun erfolgreich die Herausgabe der Zulassungsunterlagen erklagt. Tausende von Seiten warten darauf, ausgewertet zu werden. Wir werfen einen ersten Blick auf 450.000 Seiten an Dokumenten.
- Beitragsautor
Von Corona Blog Beitragsdatum 4. März 2022
Wir leben ja in einem so freien Europa, dass ganz viele Informationen, die im Zusammenhang mit den Impfstoffen und deren Zulassung stehen, nicht öffentlich gemacht werden. Dabei geht es natürlich nur um den Selbstschutz der Bürger – mit zu viel Informationen ist man heutzutage schlicht überfordert.
Die FDA wollte pro Monat lediglich ca. 500 Seiten der Dokumente herausgeben, damit wäre erst im Jahr 2096 alles einsehbar gewesen.
Klage in den USA
In den USA sah das prinzipiell nicht anders aus. Dort ist die Food and Drug Administration (FDA) für die Zulassung der Impfstoffe zuständig. Für die Zulassung des BioNTech-Pfizer Impfstoffs haben sich bei der Behörde ca. 450.000 Seiten an Dokumenten angesammelt. Bereits 2021 hatten Wissenschaftler und Forscher die Herausgabe dieser Daten nach dem „Freedom Of Information Act“ (FOIA) verlangt. Die FDA kam dem nach, allerdings wollte die Behörde pro Monat lediglich ca. 500 Seiten der Dokumente herausgeben. Damit wären erst im Jahr 2096 alle Dokumente veröffentlicht worden.
Dagegen klagten die Wissenschaftler und erst vor wenigen Wochen, am 06.01.2022, wurde darüber geurteilt: der Richter gab den Klägern recht und entschied, dass die FDA bis zum 31.01.2022 die ersten 12.000 Seiten vorlegen muss und anschließend pro weiterer 30 Tage jeweils 55.000 Seiten zu veröffentlichen hat – das erste Mal zum 01.03.2022.
Da wir die Urteilsbegründung sehr interessant finden, wollen wir einige Teile daraus übersetzen:
Wie üblich konnten sich die Parteien nicht auf einen für beide Seiten akzeptablen Zeitplan einigen, sondern legten dem Gericht zwei unterschiedliche Zeitpläne zur Prüfung vor. Dementsprechend hielt das Gericht eine Konferenz mit den Parteien ab, um einen angemessenen Veröffentlichungszeitplan festzulegen.
Eine offene Regierung ist ein grundlegend amerikanisches Thema – es ist weder ein Thema der Republikaner noch der Demokraten. Wie James Madison schrieb, ist „eine öffentliche Regierung ohne öffentliche Informationen oder die Mittel, sie zu erlangen, nur ein Anfang einer Farce oder einer Tragödie; oder vielleicht beides. Wissen wird immer über Unwissenheit herrschen: Und ein Volk, das seine eigener Herrscher sein will, muss sich mit der Macht bewaffnen, die das Wissen verleiht.“
John F. Kennedy erkannte ebenfalls, dass „eine Regierung, die Angst hat, ihr Volk in einem offenen Umfeld über Wahrheit und Lüge urteilen zu lassen, eine Regierung ist, die Angst vor ihrem Volk hat“.
Und, was in diesem Fall besonders zutreffend ist, John McCain stellte (zu Recht) fest, dass „ein übermäßiges Verwaltungsgeheimnis… Verschwörungstheorien nährt und das Vertrauen der Öffentlichkeit in die Regierung schwächt“.
In Anlehnung an diese Äußerungen besteht der grundlegende Zweck des FOIA darin, eine informierte Gesellschaft zu gewährleisten, die für das Funktionieren einer demokratischen Gesellschaft unerlässlich ist.
Der FOIA wurde [daher] erlassen, um ‚den Schleier des Verwaltungsgeheimnisses zu lüften und das Handeln der Behörde dem Licht der öffentlichen Prüfung zu öffnen‘.
Und der Kongress hat seit langem erkannt, dass ‚Informationen oft nur dann nützlich sind, wenn sie rechtzeitig vorliegen‘ und dass daher ‚eine übermäßige Verzögerung der Agentur bei ihrer Antwort oft einer Verweigerung gleichkommt‘.
[…]
In diesem Fall erkennt das Gericht die „übermäßig belastenden“ Herausforderungen an, die dieser FOIA-Antrag für die FDA darstellen kann. Aber, wie bei der Terminkonferenz zum Ausdruck gebracht, gibt es vielleicht kein wichtigeres Thema bei der Food and Drug Administration, als die Pandemie, der Impfstoff von Pfizer, die Impfung aller Amerikaner und die Gewährleistung, dass die amerikanische Öffentlichkeit sicher ist, dass dies nicht im Namen der Vereinigten Staaten überstürzt wurde.Dementsprechend kommt das Gericht zu dem Schluss, dass dieser FOIA-Antrag von überragender öffentlicher Bedeutung ist. „Schwache Informationen sind von geringem Wert“. Das Gericht stimmt mit dieser Binsenweisheit überein und kommt daher zu dem Schluss, dass die zügige Erledigung des Antrags des Klägers nicht nur durchführbar, sondern auch notwendig ist.
Urteil No. 4:21-cv-1058-P
Die Dokumente
Die Dokumente werden auf der Seite „Public Health and Medical Professionals for Transparency Documents“ für jedermann öffentlich zum Download angeboten. Wie oben beschrieben werden es noch bis Ende des Jahres monatlich ca. 55.000 Seiten mehr werden, die dazu kommen. Eine Kopie der bis zum 04.03.2022 veröffentlichten Dokumente kann auch hier heruntergeladen werden. Aktuell handelt es sich um eine Datenmenge von 324MB.
Vertrauliche Daten zu Nebenwirkungen nach bedingter Zulassung
In den letzten Tagen erhielt dabei insbesondere die Datei „5.3.6 postmarketing experience.pdf“ besondere Aufmerksamkeit. Zum Beispiel gab es dazu einen Beitrag bei TKP oder einen Twitter Post von Stefan Homburg – die unserer Meinung nach etwas irreführend formuliert sind. Man sollte trotz alledem versuchen die Sachlage so zu schildern, wie sie auch gegeben ist:
Zuerst müssen wir hier feststellen, dass die gesamte Datei (inkl. den neun Seiten ab S. 30) alles andere als neu ist. Sie wurde laut dem Zeitstempel auf PHMPT bereits am 17.11.2021 dort hochgeladen:
Eine archivierte Version der Seite vom 20.11.2021 bestätigt dies – die Datei (mit der identischen Größe) war bereits damals bei PHMPT verfügbar.
Also viel Lärm um nichts? Nein – sicher nicht. Vermutlich ging das Ganze einfach schlicht bis dato an der deutschen Öffentlichkeit vorbei (auch wir erfuhren erst durch einen Kommentar bei uns im Blog von den Dokumenten). Halten wir kurz fest: zum 01.03.2022 kam ein „ganzer Schwall“ von Dokumenten bei PHMPT dazu – aber das, worauf sich jetzt zu hauf in den sozialen Netzwerken und alternativen Medien bezogen wird, ausgerechnet das ist bereits einige Monate frei zugänglich.
Nebenwirkungen bis Februar 2021 – Deutschland sticht hervor
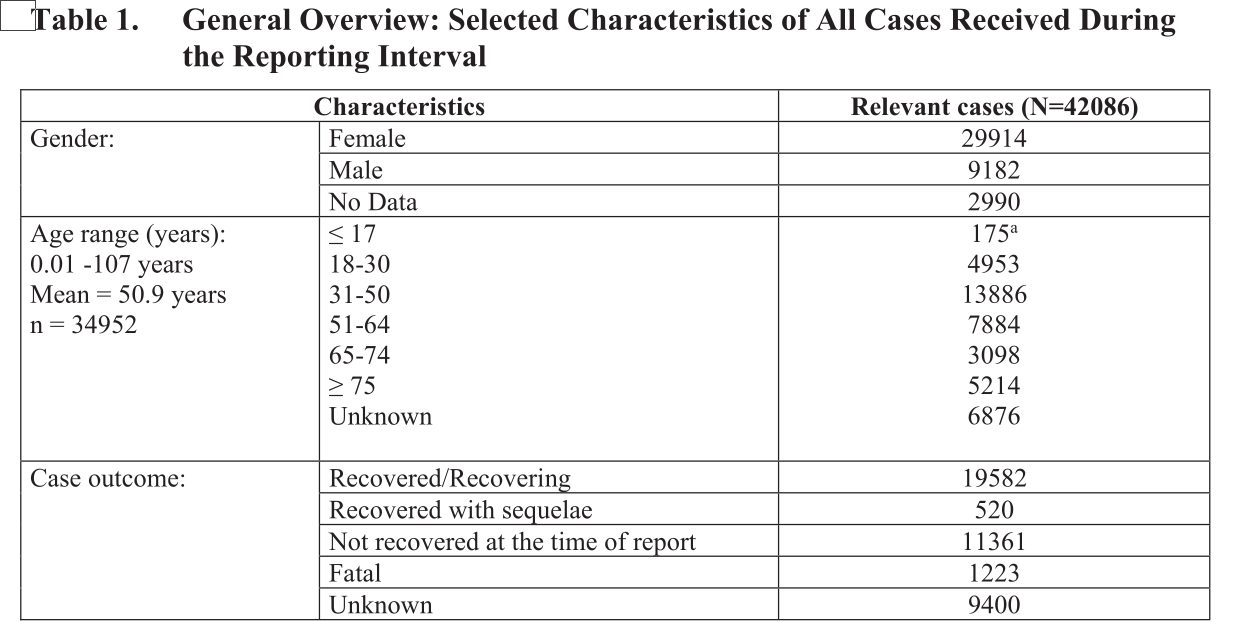
Dennoch werfen auch wir einen Blick darauf. Der Inhalt ist als „Confidential“, also vertraulich, eingestuft und hat es durchaus in sich. In dem Dokument gibt Pfizer alle Meldungen über Impfnebenwirkungen an, die im Zeitrum vom 01.12.2020 bis zum 28.02.2021 gesammelt wurden:

Hier ist zum einen interessant, dass die geschätzte Anzahl an verimpften Impfdosen in diesem Zeitraum geschwärzt wurden (das graue „(b) (4)“). So viel also zum Thema „Schwache Informationen sind von geringem Wert“…
Jedenfalls gingen in den drei Monaten ganze 42.086 Meldungen über Impfnebenwirkungen ein – die meisten (13.739) aus den USA. Aus Deutschland erhielt man 1.913 Meldungen. Die Zahl aus Deutschland ist aus einigen Gründen seltsam:
Wir vergleichen zunächst Deutschland mit dem Vereinten Königreich und Portugal. Aus dem Vereinten Königreich gingen 13.404 Meldungen ein, aus Portugal 866. Schauen wir uns demgegenüber die Anzahl der verimpften Dosen in diesen Ländern Ende Februar 2021 an:
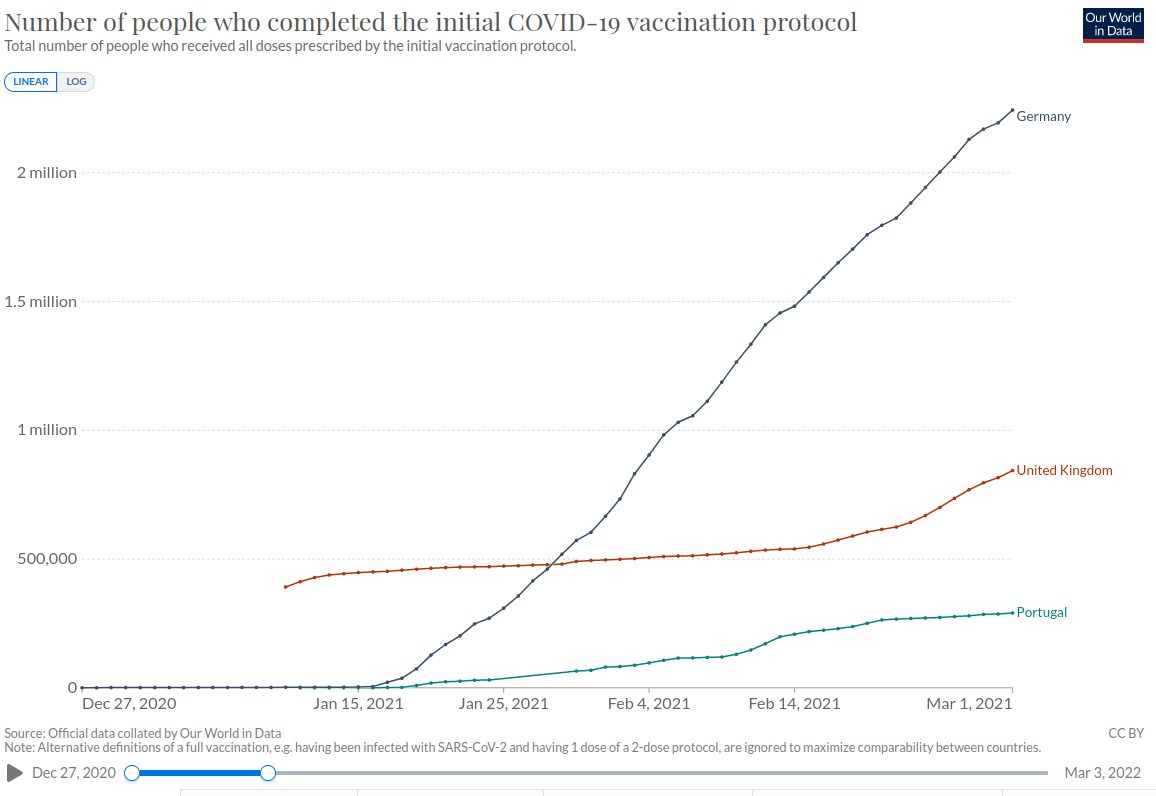
Am 28.02.2021 waren in Deutschland demnach 2.190.000 Menschen mindestens ein Mal geimpft. Im Vereinten Königreich waren es 815.816 Menschen und in Portugal 286.470.
Wir haben also pro 1.000 Impfungen in Deutschland 0,9 Nebenwirkungen. In Portugal sind es 3,0 Nebenwirkungen und im Vereinten Königreich 16,4 Nebenwirkungen pro 1.000 Impfungen.
Deutschland ist eindeutig „Spitzenreiter“ – entweder die Deutschen vertragen die Impfung besser, als ihre europäischen Landsmänner, oder… naja, über die Dunkelziffer haben wir ja schon so einiges gesagt.
Weiterhin fällt auf, dass Deutschland bis zum 28.02.2021 gerade einmal 1.913 Nebenwirkungen gemeldet hat. Das ist eine extreme Diskrepanz, zu den Angaben im entsprechenden Sicherheitsbericht des Bundesinstituts für Impfstoffe und biomedizinische Arzneimittel (Paul-Ehrlich-Institut, PEI) aus diesem Zeitraum. Bis zum 26.02.2021 gingen beim PEI nämlich 11.915 Meldungen über Impfnebenwirkungen ein. Uns klingen da noch die Worte aus dem 17. Sicherheitsbericht des PEI in den Ohren:
Das Paul-Ehrlich-Institut ist nach dem Arzneimittelgesetz verpflichtet, Meldungen zu Verdachtsfällen von Nebenwirkungen in bestimmten Zeitabständen elektronisch in einem international einheitlichen Format und pseudonymisiert an die gemeinsam EudraVigilance-Datenbank bei der Europäischen Arzneimittelagentur zu melden, zu der jede Zulassungsbehörde in der EU Zugang hat.
PEI im 17. Sicherheitsbericht
Das PEI übermittelt offensichtlich die Daten – nur halt mit Wochen oder gar Monaten Verzögerung. Ein wahres Armutszeugnis.
Das Ganze im Detail
In dem Dokument wird ferner in einer Abbildung die grobe Altersverteilung der Fälle und der Ausgang der Reaktionen angegeben:
Hier hat uns überrascht, dass im Februar 2021, zu einer Zeit, als zumindest in Deutschland mit Masse die Senioren geimpft wurden, die meisten Nebenwirkungen in der Gruppe der 31 – 50 Jährigen vermeldet wurde (13.886 Fälle). Auch Frauen waren deutlich überrepräsentiert (29.914). Das PEI gibt die Verteilung der Nebenwirkungen auf die Geschlechter mittlerweile gar nicht mehr an. Zum letzten Mal wurden hier im 14. Sicherheitsbericht Daten vom PEI veröffentlicht, die ebenfalls eine starke Überrepräsentation von Frauen zeigten.
Von den 42.086 eingegangenen Fällen endeten 1.223 tödlich („Fatal“) – ganze 2,9%. Eine wirklich „ordentliche Quote“. Rechnet man die 9.400 Fälle mit unbekanntem Ausgang heraus, dann endeten 3,7% der Fälle mit bekanntem Ausgang tödlich.
Eine weitere Grafik in dem Dokument zeigt die Art der Nebenwirkung und ob sie „normal“ (blau) oder schwerwiegend (violett) war:
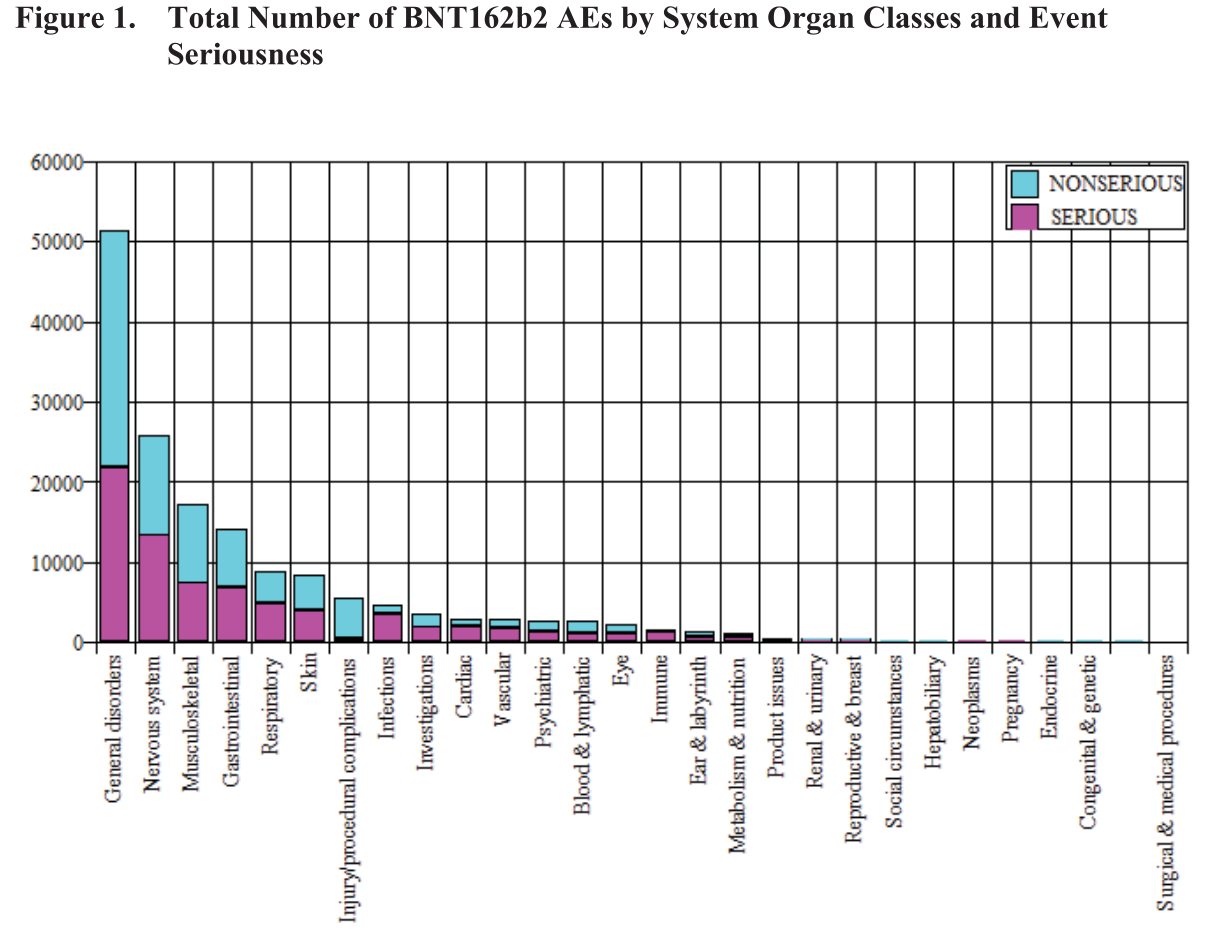
Ferner sind in einer weiteren Tabelle einige Reaktionen aufgelistet, die in mehr als 2% der Fälle gemeldet wurden, z.B.:
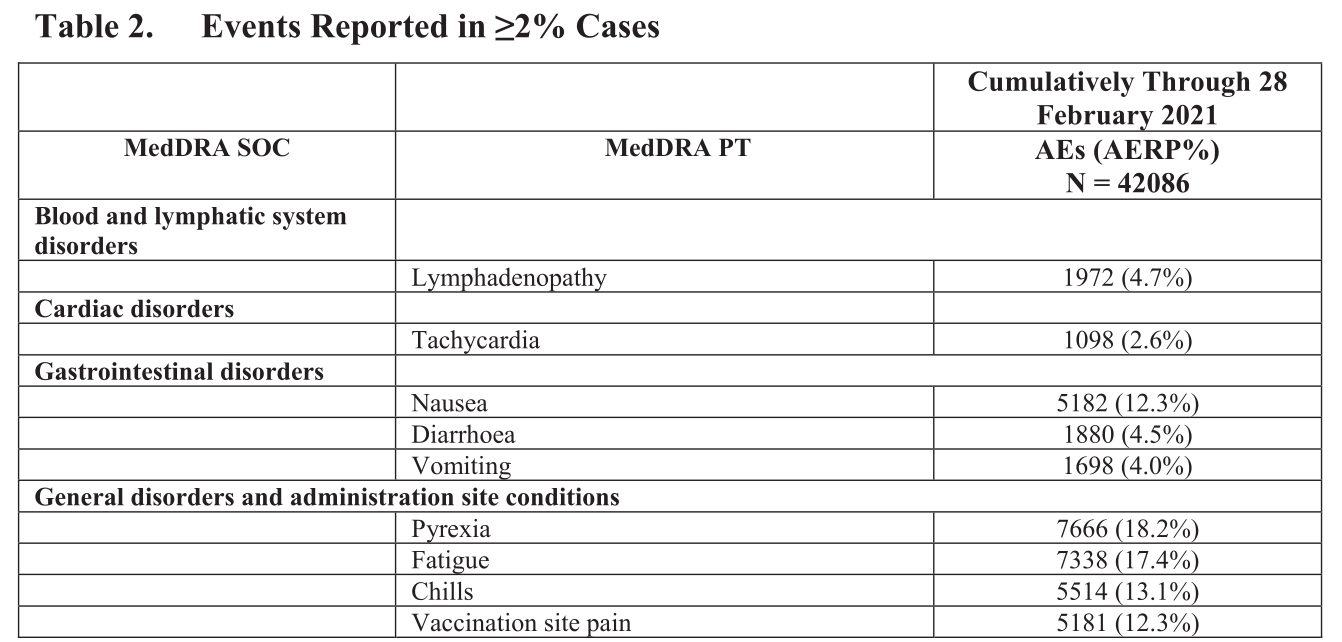
Also Probleme mit dem Herzen waren schon nach 3 Monaten in der „Hitliste“ der Impfnebenwirkungen.
Vorsicht vor Fehlinformationen: 9 Seiten mit Diagnosen zu Impfnebenwirkungen?
Wir hatten oben schon ein Twitter Zitat von Stefan Homburg gebracht, in dem er auf 9 Seiten an Nebenwirkungen ab S. 30 in dem Dokument eingeht. In den sozialen Netzwerken ging das Ganze auch teilweise „viral“. Hier nur ein Auszug einer halben Seite (von den insgesamt neun):

Wir haben also unzählige Symptome oder Erkrankungen, die jeweils mit einem „;“ voneinander getrennt sind.
Doch Vorsicht, auf S. 16 im Dokument steht eine nähere Erläuterung zu dieser Liste, hier in Übersetzung:
3.1.3. Überprüfung der unerwünschten Ereignisse von besonderem Interesse (AESI)
Die Liste der AESI des Unternehmens für BNT162b2 finden Sie in Anhang 1.
Die AESI-Liste des Unternehmens berücksichtigt die Listen der AESI der folgenden Expertengruppen und Zulassungsbehörden: Brighton Collaboration (SPEAC), ACCESS-Protokoll, US CDC (vorläufige Liste der AESI für die VAERS-Überwachung), MHRA (unveröffentlichter Leitfaden).
Die AESI-Begriffe werden in eine TME-Liste [Targeted Medically Event, gezieltes medizinisches Ereignis] aufgenommen und umfassen Ereignisse, die aufgrund ihres Zusammenhangs mit einer schweren COVID-19 Erkrankung von Interesse sind, sowie Ereignisse, die für Impfstoffe im Allgemeinen von Interesse sind.
Die AESI-Liste besteht aus MedDRA-PTs, HLTs, HLGTs oder MedDRA-SMQs und kann je nach dem sich entwickelnden Sicherheitsprofil des Impfstoffs geändert werden.Tabelle 7 gibt einen zusammenfassenden Überblick über die kumulativen Fälle innerhalb der AESI-Kategorien in der Sicherheitsdatenbank von Pfizer. Dies unterscheidet sich von der Bewertung von Sicherheitssignalen, die durchgeführt und gegebenenfalls in die monatlichen Sicherheitsberichte aufgenommen werden, die regelmäßig der FDA und anderen Gesundheitsbehörden vorgelegt werden.
Die neun seitige Liste enthält also lediglich eine Auflistung aller Symptome, die überwacht werden und die aus verschiedenen anderen Listen übernommen wurden.
Spannender ist da schon Tabelle 7, wo dann gezielt die Symptome aufgeführt werden, die tatsächlich aufgetreten sind, hier ein Auszug daraus:

Ende Februar 2021 gab es also 1.403 Fälle mit Bezug zu Herzproblemen. 136 davon endeten tödlich. Das Fazit könnte auch aus einem deutschen PEI Sicherheitsbericht stammen:
Schlussfolgerung: Diese kumulative Fallprüfung wirft keine neuen Sicherheitsfragen auf. Die Überwachung wird fortgesetzt.
Weitere Details zu Nebenwirkungen bei den Zulassungsstudien
In dem Dokument STN 125742_0_0 Section 2.5 Clinical Overview.pdf finden sich unzählige Details zu den Nebenwirkungen während der ersten Zulassungsstudien. Interessant dabei ist:
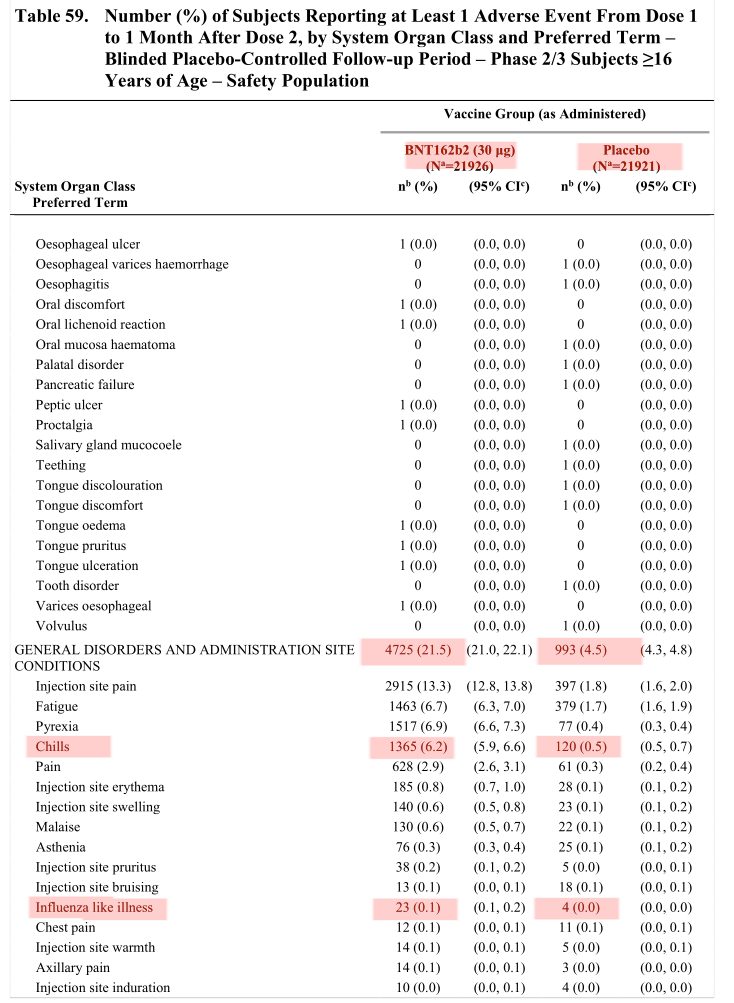
Wir haben also über 11 Mal so viele Fälle von Fieber nach der Impfung als nach dem Placebo und knapp 6 Mal so viele Fälle von „grippeähnlichen Symptomen“ nach der Impfung als nach Placebo.
Im Dokument 125742_S1_M1_priority-review-request.pdf finden sich Details zu lokalen Reaktionen während einer Studie nach der Impfung. Hier ist auffällig, dass bei „Rötungen, Schwellungen und Schmerzen an der Einstichstelle“ die „schweren Fälle“ in der Gruppe der Geimpften immer deutlich über denen in der Placebogruppe sind (da im Originaldokument rot, haben wir es in grün hervorgehoben):
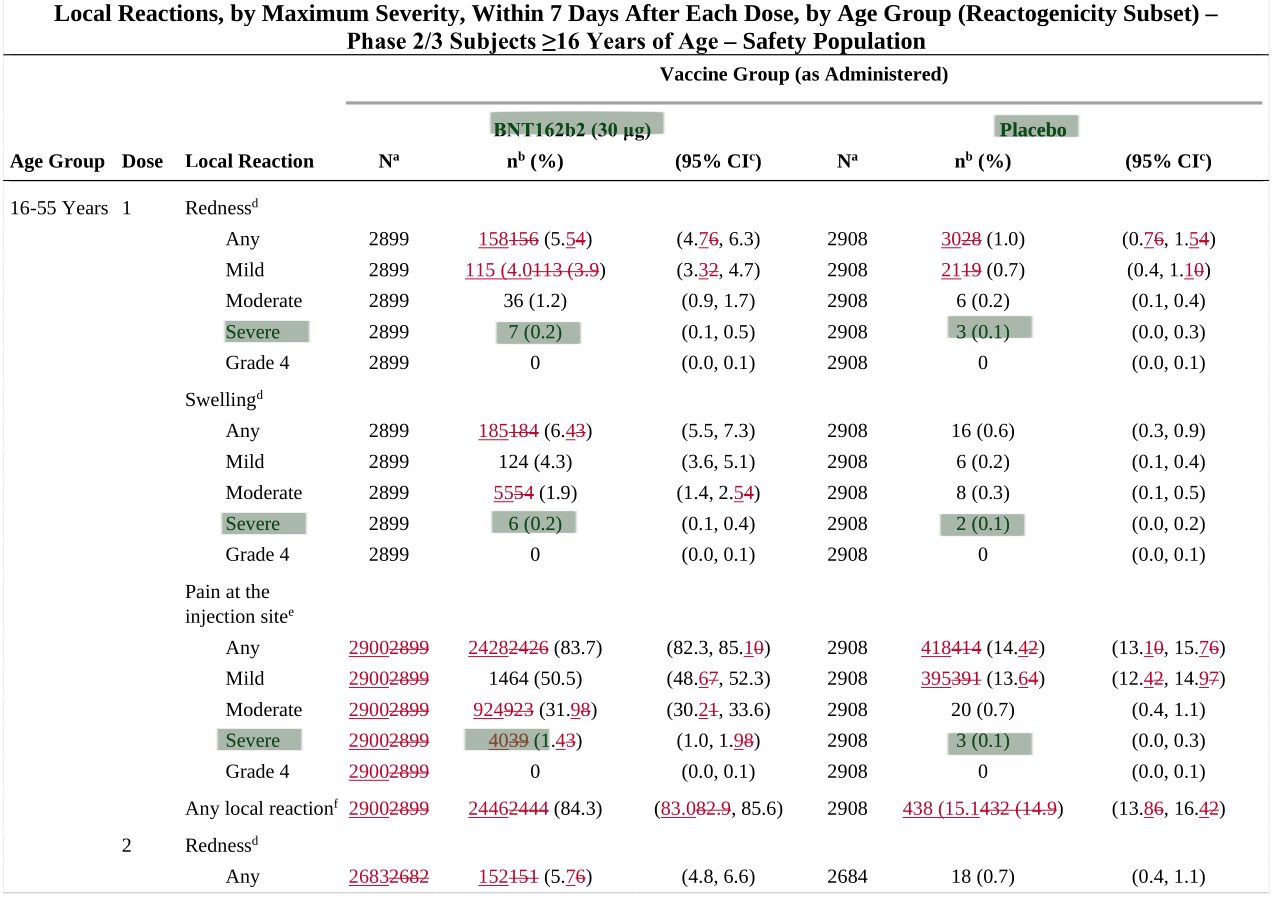
Was immer man von einem „schweren Verlauf“ bei einer „Rötung“ verstehen mag. Jedenfalls sind in allen Kategorien die BioNTech-Pfizer Probanden deutlich vor der Placebogruppe. Wieder einmal beschleicht uns das Gefühl, dass man sprichwörtlich den Teufel mit dem Belzebub austreiben möchte.
Verantwortliche bei den Zulassungsstudien
In den Dokumenten finden sich auch alle Einrichtungen aufgelistet, die bei der Zulassungsstudie des Impfstoffs mitgewirkt haben. Mit aufgeführt sind auch immer die verantwortlichen Personen in diesen Einrichtungen, zum Beispiel für Deutschland:
Lustigerweise sind zu allen Ansprechpersonen auch die „Bewerbungsschreiben“ für die Teilnahme an der Zulassungsstudie (in einer anderen Datei) mit dabei. Beispielsweise hier ein Auszug für Antje Blank:

Ach und wer von unseren Lesern einmal in die Verlegenheit geraten sollte, ein paar „Lücken“ im Lebenslauf überbrücken zu müssen, dem empfehlen wir die Methode von Frau Blank, das Zauberwort heißt „Family management“:

Die Sache mit der Verteilung der LNP im Körper
Ein brandneues (als vertraulich eingestuftes) Dokument vom 01.03.2022 ist 125742_S1_M2_24_nonclinical-overview.pdf. In diesem geht es um die Ausbreitung der Lipidnanopartikel (LNP) im Körper von Tieren, insbesondere Ratten. Wie wir erst kürzlich berichteten, greift eine neue Studie aus Schweden auch dieses Thema auf und zeigt, dass die mRNA des Impfstoffs in Organen wie der Leber in DNA umgewandelt werden kann. Diese hat zumindest das Potenzial, das Erbgut des Menschen zu verändern und zur Entstehung von Krebs beizutragen. In dem neu veröffentlichten Dokument heißt es zum Thema „Karzinogenität“:
Mit BNT162b2 wurden keine Karzinogenitätsstudien durchgeführt, da es sich bei den Bestandteilen des Impfstoffkonstrukts um Lipide und RNA handelt und von ihnen kein karzinogenes oder tumorerzeugendes Potenzial erwartet wird.
Das ist doch eine saubere, wissenschaftliche Begründung. Man erwartet etwas nicht und in freudiger Erwartung spritzt man es dann mal Millionen von Menschen. Hoffen wir, dass sich die Erwartungen von BioNTech-Pfizer erfüllen und die umgewandelte RNA wirklich nicht ins Erbgut des Menschen übergehen kann.
In dem besagten Dokument werden dann die Versuche an Ratten näher beschrieben, die die Ausbreitung der LNP in den Tieren untersuchten. Unter anderem wird die Konzentration der Stoffe ALC-0315 und ALC-0159 in Plasma und Leber für einen gewissen Zeitraum nach der Injektion aufgetragen:
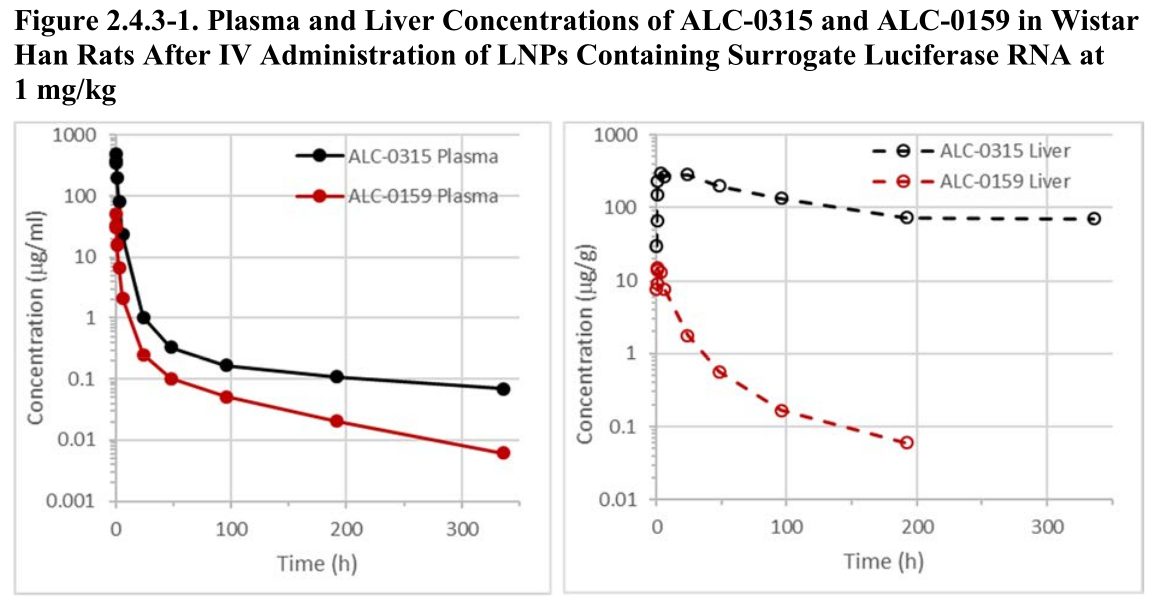
Das sind übrigens genau die Stoffe, die einige Professoren in Deutschland in einem offenen Brief an BioNTech Vorstand Ugur Sahin ansprachen und die ihnen Sorgen bereiteten.
In dem Dokument heißt es außerdem:
In der Studie an Ratten wurde nach intravenöser Verabreichung von LNPs […] keine nachweisbare Ausscheidung von ALC-0315 und ALC-0159 im Urin festgestellt. Der Prozentsatz, der unverändert mit den Fäkalien ausgeschieden wurde, betrug ~1% für ALC-0315 und ~50% für ALC-0159. Metaboliten von ALC-0315 wurden im Urin von Ratten nachgewiesen (Abbildung 2.4.3-3).
Die Abbildung 2.4.3-3 zeigt dann den „Vorgeschlagenen Biotransformationsweg von ALC-0315 in verschiedenen Spezies“ – insbesondere das Wort „Vorgeschlagen“ klingt für uns als alles andere, als einer abgeschlossenen Wissenschaft. Man beachte, wo der Stoff (oder Umwandlungsprodukte davon) überall landen (Blut, Leber, Plasma):
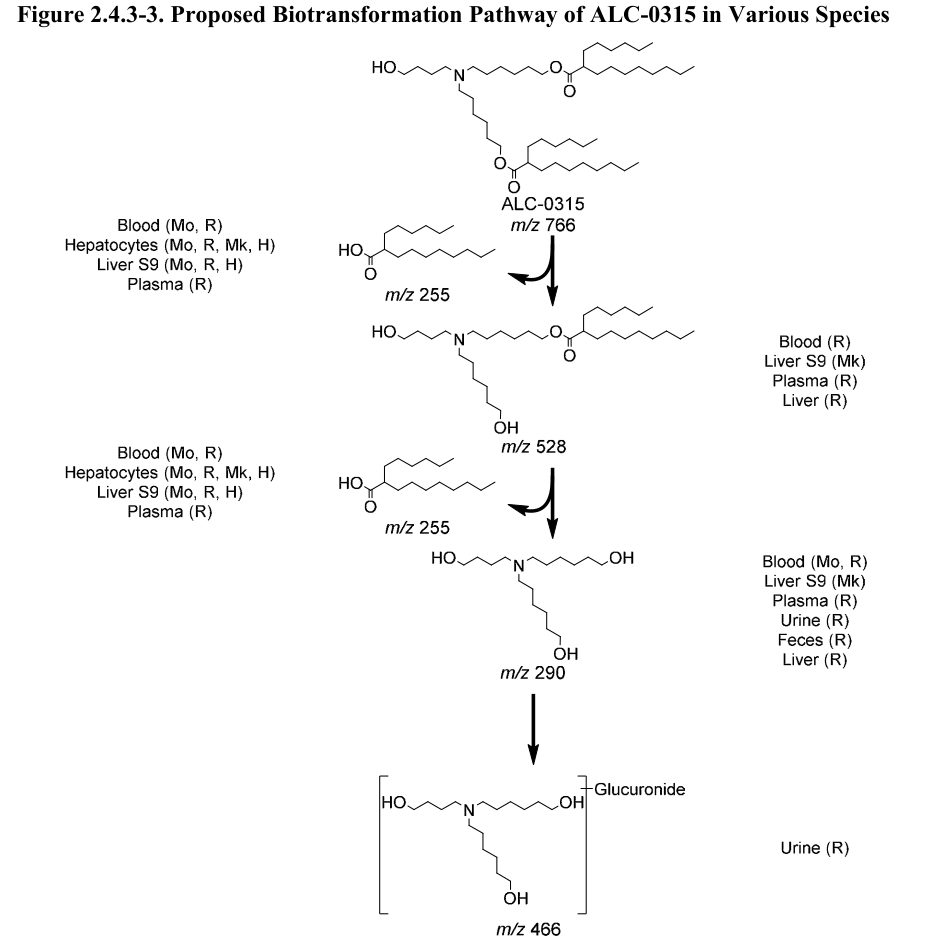
Interessant ist auch, dass das nun ausführlichere Informationen zu einem Thema sind, die schon einmal im Zuge der EMA-Leaks an die Öffentlichkeit gelangten. Ein Wissenschaftler hatte uns schon im Januar 2021 aus den EMA-Leaks folgendes ausgewertet:
In Anbetracht der Tatsache, dass Acetamine in Tierversuchen als karzinogen, einschließlich Lebertumoren, eingestuft wurden, was möglicherweise auf einem genotoxischen Mechanismus beruht, wird der Antragsteller gebeten, eine Diskussion über die Verteilung und den Metabolismus von ALC-0159 mit Schwerpunkt auf dem cetamid-Anteil zu geben.
Da fast kein unverändertes ALC-3015 im Urin oder in den Fäkalien nachgewiesen wurde, könnte der Metabolismus eine größere Rolle bei der Eliminierung von ALC-0315 als von ALC-0159 spielen.
Der Antragsteller wird gebeten, das Risiko, dass der mRNA-Impfstoff potenzielle Autoimmunreaktionen auslösen kann, weiter zu erörtern und darzulegen, wie er plant, deren Auftreten möglicherweise zu bewerten.
Wir sind auf diesem Gebiet selbst keine Fachleute und sind wirklich gespannt, was Experten dazu sagen können bzw. was weitere Studien dazu ergeben.
Fazit
Wir konnten in dem Artikel hier nur einen wirklich kurzen Überblick über die veröffentlichten Dokumente geben. Der Grund ist ganz einfach: es ist für uns schlicht unmöglich, die tausenden von Seiten im Detail auszuwerten.
Wir können zumindest sagen, dass unser „schlechtes Bauchgefühl“ bei den Daten und Auswertungen des PEI verstärkt wurde und dass das, was wir gelesen haben, nach alles andere als einem „gut erprobten Impfstoff“ oder einem „vollständig verstandenem Wirkprinzip“ klingt.
Quelle



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hinterlasse einen Kommentar
Du musst angemeldet sein, um einen Kommentar schreiben zu können.